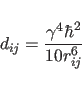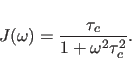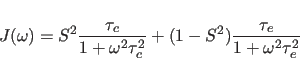|
| Xplor-NIH home Documentation |
Next: Analytical Expression for the Up: NMR Back-calculation Refinement Previous: Output
|
| Xplor-NIH home Documentation |
![\begin{displaymath}
I_{ij}^{c} \propto [\exp(-{\bf R} \tau_{m})]_{ij},
\end{displaymath}](img802.png) |
(39.1) |
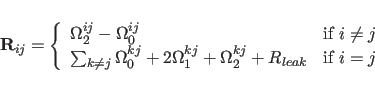 |
(39.2) |
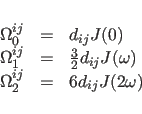 |
(39.3) |
In the simplest model, it is assumed that a single
isotropic correlation time ![]() is
sufficient to describe the spectral densities
is
sufficient to describe the spectral densities ![]() (Solomon, 1955):
(Solomon, 1955):
A step beyond this simple model is the “model-free" approach of
Lipari and Szabo (1982), where the internal motion is described by two
parameters,
an effective correlation time ![]() and an order parameter
and an order parameter ![]() :
:
Groups of protons whose resonances are degenerate due to motion
(in general, mostly methyl groups) are
treated roughly as in
CORMA, version 1.5 (Keepers and James 1984).
(Note that cross peaks which are ambiguous due to overlap should be
dealt with in a different way; see the example input file
in Section 39.7.)
Each such group
is represented by
one spin, whose intensity is scaled by the number of protons in the group,
and the distance to
the group
is calculated as the
![]() or
or
![]() average over
the protons in the group (Eq. 39.4).
In addition, a diagonal leakage rate is added for each group
of protons.
average over
the protons in the group (Eq. 39.4).
In addition, a diagonal leakage rate is added for each group
of protons.
Protons can be removed from the spin system (exchangeable protons in
D![]() O spectra, deuterium-labeled molecules), or their appropriate
occupancy can be specified
(exchangeable protons in H
O spectra, deuterium-labeled molecules), or their appropriate
occupancy can be specified
(exchangeable protons in H![]() O spectra).
O spectra).