
| bit | Xplor-NIH | VMD-XPLOR |
|---|
|
| Xplor-NIH home Documentation |
Next: Requirements Up: Merging Trajectories Previous: Merging Trajectories
Syntax
- DYNAmics MERGe
- {
 dynamics-merge-statement
dynamics-merge-statement } END
is invoked from the main level of X-PLOR
} END
is invoked from the main level of X-PLOR
- dynamics-merge-statement :==
-
- trajectory-statement
- is defined in Section 12.1.3.
- ENSEmble=logical
- for ENSEmble=TRUE bypasses the mismatch checking between subsequent trajectory files. In this way it is possible to generate a pseudotrajectory from a group of otherwise unrelated trajectory files for the same system. One example where this option is useful consists of the merging of several independent molecular dynamics simulations in order to compute time-independent properties.
- FORMat=string
- is the FORTRAN format
for the OASCii=TRUE option.
If the hexadecimal format is specified when writing an
ASCII trajectory file, care should be taken to ensure that the
coordinates after application of the
scale factor
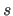 and the offset
and the offset 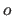 are all positive during
the whole course of the molecular dynamics simulation.
Otherwise, FORTRAN format runtime errors will occur
during writing or reading of the ASCII file. This is
of particular concern when one is writing velocities. The
default format and scale work well for Cartesian
coordinates but do not work for velocities
(default=12Z.6, hexadecimal).
are all positive during
the whole course of the molecular dynamics simulation.
Otherwise, FORTRAN format runtime errors will occur
during writing or reading of the ASCII file. This is
of particular concern when one is writing velocities. The
default format and scale work well for Cartesian
coordinates but do not work for velocities
(default=12Z.6, hexadecimal).
- OASCii=logical
- writes a formatted (ASCII) file if
OASCii is TRUE; if OASCii
is FALSE, an unformatted (binary) file is written.
A scale factor and an offset are applied to the
coordinates
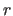 before they are written to an ASCII file; i.e.,
before they are written to an ASCII file; i.e.,
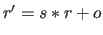 .
.
- OFFSet=real
- provides an offset for the OASCii= TRUE
option (default: 800).
- ORIEnt { orient-statement } END
- fits the input trajectory frames to the main coordinate set using a least-squares fitting procedure and then writes the frames to the trajectory output file.
- OUTPut=filename
- designates an output file for the trajectory (default: OUTPUT).
- SCALe=real
- provides a scale factor for the
OASCii=TRUE option (default: 10000).
- orient-statement:==
-
- MASSweighting=logical
- is a logical flag indicating whether mass-weighting
is applied for the least-squares fitting
procedure (default:
FALSE). - NOROtation=logical
- is a logical flag indicating whether a rotation is applied to the coordinates. If FALSE, only a translation vector is applied (default: TRUE).
- SELEction=selection
- specifies the atoms that are used to define the least-squares transformation. However, the transformation is applied to all coordinates (default: (ALL) ).
- WEIGhting-with-B=logical
- is a logical flag indicating whether the B-array is used for weighting the atoms for the least-squares fitting procedure (default: FALSE).
- MASSweighting=
Xplor-NIH 2026-07-10