
| bit | Xplor-NIH | VMD-XPLOR |
|---|
|
| Xplor-NIH home Documentation |
Next: Requirements Up: Electron Density Maps Previous: Electron Density Maps
Syntax
- MAP
- {
 xrefin-map-statement
xrefin-map-statement } END
is an xrefin statement. Action takes place as soon as the END
statement is issued.
} END
is an xrefin statement. Action takes place as soon as the END
statement is issued.
- xrefin-map-statement:==
-
- AUTOmatic=logical
- is a flag indicating that
automatic scaling is performed. The electron density map
will have an average of zero and a standard deviation (
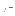 ) of one
(default: TRUE).
) of one
(default: TRUE).
- CUSHion=real
- specifies additional cushioning around the molecule in Å. Only for EXTEnd=MOLEcule mode (default: 2.0 Å).
- EXTEnd=MOLEcule
 UNIT BOX
UNIT BOX - is a mode for boundary specification (default: MOLEcule). In MOLEcule mode, the map covers the selected atoms of the molecule. In UNIT mode, the whole unit cell is written. In BOX mode, the user can specify the boundaries of a cubic box in orthogonal Å.
- FORMatted=logical
- is a flag that indicates whether a formatted (FORM=TRUE) or binary (FORM=FALSE) file is written (default: TRUE).
- IOUTput=filename
- is a filename for the imaginary portion of the electron density map; it works only when HERMitian is set to FALSE (cf. Section 13.3) (default: no output).
- OUTPut=filename
- is the filename for the real portion of the electron density map (default: standard output).
- SELEction=selection
- selects atoms for EXTEnd=MOLEcule mode. This selection is independent from the selection in the xrefin statement. The map is written to cover the selected atoms (default: all).
- XMAX
- is only for EXTEnd=BOX mode; it specifies upper x boundary of box in orthogonal Å (default: 1.0).
- XMIN
- is only for EXTEnd=BOX mode; it specifies lower x boundary of box in orthogonal Å (default: 0.0).
- YMAX
- is only for EXTEnd=BOX mode; it specifies upper y boundary of box in orthogonal Å (default: 1.0).
- YMIN
- is only for EXTEnd=BOX mode; it specifies lower y boundary of box in orthogonal Å (default: 0.0).
- ZMAX
- is only for EXTEnd=BOX mode; it specifies upper z boundary of box in orthogonal Å (default: 1.0).
- ZMIN
- is only for EXTEnd=BOX mode; it specifies lower z boundary of box in orthogonal Å (default: 0.0).
- AUTOmatic=