
| bit | Xplor-NIH | VMD-XPLOR |
|---|
|
| Xplor-NIH home Documentation |
Next: Requirements Up: Parameter Statement Previous: Parameter Statement
Syntax
- PARAmeter {
 parameter-statement
parameter-statement } END is
invoked from the main level of X-PLOR.
} END is
invoked from the main level of X-PLOR.
- parameter-statement:==
-
- ANGLe
- type type type real
real [UB real real]
adds a bond angle parameter set for the three atom
types to the parameter database.
The first real specifies
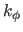 , which has units of
kcal mole
, which has units of
kcal mole rad
rad , and
the second real specifies
, and
the second real specifies 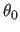 , the
equilibrium angle,
which has units of degrees (Eq. 4.5).
The optional UB specification activates the Urey-Bradley
term (Eq. 4.5), where the first real is the
Urey-Bradley energy constant
, the
equilibrium angle,
which has units of degrees (Eq. 4.5).
The optional UB specification activates the Urey-Bradley
term (Eq. 4.5), where the first real is the
Urey-Bradley energy constant 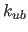 and the second real
is the Urey-Bradley equilibrium distance
and the second real
is the Urey-Bradley equilibrium distance 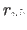 between the first
and the third atom that define the angle. If UB is not specified,
the Urey-Bradley equilibrium distance and energy constant default
to zero.
The program automatically
performs an interchange of the first with the third atom types
where this is required.
between the first
and the third atom that define the angle. If UB is not specified,
the Urey-Bradley equilibrium distance and energy constant default
to zero.
The program automatically
performs an interchange of the first with the third atom types
where this is required.
- ANGLe
- selection
selection selection
real real [UB real real]
is an atom-based version of the ANGLe statement that
specifies the bond angle parameters to be used for any angles in the
molecular structure that match the given triple atom selection.
It can apply to more than one angle, depending on the number
of angles that match the triple atom selection.
The definition of the
reals is identical to the type-based angle statement.
The energy and equilibrium constants can
be set to the string TOKEn, which
implies that the corresponding parameter
will be untouched by this statement.
- BOND
- type type real real
adds
a covalent bond parameter set for the two atom
types to the parameter database.
The first real specifies
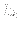 , which is the energy
constant in units of kcal mole Å, and
the second real specifies
, which is the energy
constant in units of kcal mole Å, and
the second real specifies
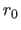 , which is the equilibrium bond length in Å
(Eq. 4.4).
The program automatically performs
an interchange of the two atom types
where this is required.
, which is the equilibrium bond length in Å
(Eq. 4.4).
The program automatically performs
an interchange of the two atom types
where this is required.
- BOND
- selection selection real real
is an atom-based version of the BOND statement. The definition of the
reals is identical to the type-based bond statement.
It can apply to more than one bond, depending on the number
of bonds that match the double atom selection.
The energy and equilibrium constants can
be set to the string TOKEn, which
implies that the corresponding parameter
will be untouched by this statement.
- DIHEdral
- type type type type
[MULT integer] {real integer real }
adds a dihedral angle parameter set for the
four atom types to the parameter database
(see also Eq. 4.6).
The MULT option specifies the multiplicity
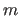 of the dihedral
angle (default: m=1). For multiple dihedrals of multiplicity
, there are groups of
of the dihedral
angle (default: m=1). For multiple dihedrals of multiplicity
, there are groups of 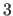 items
following the MULT integer statement.
The first real of each group specifies
items
following the MULT integer statement.
The first real of each group specifies 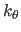 , the
integer is the periodicity
, the
integer is the periodicity 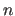 , and the
second real specifies
, and the
second real specifies 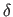 , the phase-shift angle, which
has units
of degrees. If the periodicity is greater than 0,
, the phase-shift angle, which
has units
of degrees. If the periodicity is greater than 0, 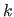 has the
units of kcal mole; if the periodicity is 0, has the units
of kcal mol rad (Eq. 4.6).
The special character X is reserved for
the following combination: X type type X; it
acts as a wildcard. The
parameter retrieval
for a specified dihedral angle proceeds in the following way: first, a match
without wildcards is attempted, and second, a match against dihedral
parameters containing
wildcards is attempted. Some instances (such as sugars) require the mixing of
multiple dihedral angle terms with different periodicities.
In this case, dihedral statements with the multiple option
should be given in the parameter statement and
in the topology statement (Section 3.1.1).
Wildcards are
not allowed for multiple dihedral angles. The program automatically
performs the interchange
(a b c d)
has the
units of kcal mole; if the periodicity is 0, has the units
of kcal mol rad (Eq. 4.6).
The special character X is reserved for
the following combination: X type type X; it
acts as a wildcard. The
parameter retrieval
for a specified dihedral angle proceeds in the following way: first, a match
without wildcards is attempted, and second, a match against dihedral
parameters containing
wildcards is attempted. Some instances (such as sugars) require the mixing of
multiple dihedral angle terms with different periodicities.
In this case, dihedral statements with the multiple option
should be given in the parameter statement and
in the topology statement (Section 3.1.1).
Wildcards are
not allowed for multiple dihedral angles. The program automatically
performs the interchange
(a b c d) 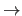 (d c b a) where this is required.
(d c b a) where this is required.
- DIHEdral
- selection selection selection
selection
[MULTinteger]
{ real integer real }
is an atom-based version of the DIHEdral statement.
The
definition of the real and integer numbers is identical
to the DIHEdral definition (see also Eq. 4.6).
For multiple dihedrals of multiplicity
, there are 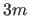 items following the MULT integer statement
(i.e., sets of one energy constant, one periodicity, and one offset).
The statement can apply to more than one dihedral angle, depending
on the number
of bonds that match the quadruple atom selection.
An atom-based statement takes priority over any
type-based parameter that may have been specified earlier.
The energy constants, periodicities, and offsets can
be set to the string TOKEn, which
implies that the corresponding constant
will be untouched by this statement.
items following the MULT integer statement
(i.e., sets of one energy constant, one periodicity, and one offset).
The statement can apply to more than one dihedral angle, depending
on the number
of bonds that match the quadruple atom selection.
An atom-based statement takes priority over any
type-based parameter that may have been specified earlier.
The energy constants, periodicities, and offsets can
be set to the string TOKEn, which
implies that the corresponding constant
will be untouched by this statement.
- HBONded
- *type* *type* real real
adds an explicit hydrogen-bonding parameter set for the
specified pair of atom types to the parameter database.
The first atom type refers to donor (heavy) atoms, whereas the second
one refers to the acceptor atoms. Wildcards are allowed for
both donor and acceptor atom types.
The first real is the well depth
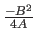 and the second is the distance
and the second is the distance
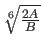 (Eq. 4.25).
(Eq. 4.25).
- HBONDS
- { hbonds-statement } END
specifies global hydrogen-bond
parameters for the explicit hydrogen-bond energy term (see Eq. 4.25).
- IMPRoper
- type type type type
[MULT integer] {real integer real }
adds an improper angle parameter set for the
four atom types to the parameter database.
The definition is identical to the DIHEdral definition
(see also Eq. 4.6),
except for wildcards.
The wildcard cascading for improper angles is as follows:
(a b c d) (a X X d) (X b c d)
(X b c X) (X X c d).
The program automatically
performs the interchange
(a b c d) (d c b a) where this is required.
- IMPRoper
- selection selection selection
selection
[MULTinteger]
{ real integer real }
is an atom-based version of the IMPRoper statement. The
definition of the real and integer numbers is identical
to the DIHEdral definition (see also Eq. 4.6).
The statement can apply to more than one improper angle, depending
on the number
of bonds that match the quadruple atom selection.
The energy constants, periodicities, and offsets can
be set to the string TOKEn, which
implies that the corresponding constant
will be untouched by this statement.
- LEARn learn-statement END
- learns atom-based parameters from one or more sets of
Cartesian coordinates (see Section 3.4).
- NBFIx
- type type real
real real real
adds a Lennard-Jones parameter set for the specified pair of atom
types to the parameter database.
The first two real numbers are the A, B coefficients
(Eqs. 4.12
and 4.13) for all nonbonded interactions
except the special 1-4 interactions; the
second pair of reals is for
the 1-4 (NBXMod=
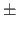 5) nonbonded interactions.
Appropriate NONB statements have to be specified
for both atom types
before invoking this statement. The NBFIx statement allows
one to deviate from the standard combination rule for the
Lennard-Jones potential (Eq. 4.14).
5) nonbonded interactions.
Appropriate NONB statements have to be specified
for both atom types
before invoking this statement. The NBFIx statement allows
one to deviate from the standard combination rule for the
Lennard-Jones potential (Eq. 4.14).
- NBFIx
- selection selection
real real real real
is an atom-based version of the NBFIx statement that
adds a Lennard-Jones parameter set for the selected pairs of atoms
to the atom-based parameters. The definition of the
reals is identical to the type-based NBFIx statement.
Appropriate atom-based
NONB statements have to be specified for both atom
selections
before invoking this statement, e.g.,
NONB ( chemical C* ) 0.12 3.74 0.12 3.74 NONB ( chemical N* ) 0.24 2.85 0.24 2.85 NBFIx ( chemical C* ) ( chemical N* ) 10. 1000. 10. 1000.
Note that the TOKEn keyword is not allowed for the reals. - NBONds { nbonds-statement } END
- applies to both electrostatic and van der Waals energy calculations. It sets up global parameters for the nonbonded interaction list generation and determines the form of subsequent nonbonded energy calculations (see Eq. 4.8).
- NONB
- type real real real real
adds a Lennard-Jones parameter set for pairs of atoms of the same
specified type to the parameter database.
The first pair of reals is
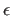 ,
,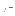 (Eq. 4.8)
for all nonbonded interactions except the
special 1-4 interactions; the second pair
is , for the 1-4 nonbonded
interactions (NBXMod=5).
(Eq. 4.8)
for all nonbonded interactions except the
special 1-4 interactions; the second pair
is , for the 1-4 nonbonded
interactions (NBXMod=5).
- NONB
- selection
real real real real
is an atom-based version of the NONB statement that
adds a Lennard-Jones parameter set for the selected atoms
to the atom-based parameters. The definition of the reals is
identical to the type-based NONB statement. Note that
the TOKEn keyword is not allowed for the reals.
- REDUce { reduce-statement } END
- derives type-based parameters from existing atom-based parameters
(see Section 3.5).
- RESEt
- [ ALL
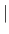 TYPE ATOM ] erases all information about
type- and atom-based parameters. The optional specification of ALL erases
both databases, TYPE erases just the type base, and ATOM erases just
the atom base (default: ALL).
TYPE ATOM ] erases all information about
type- and atom-based parameters. The optional specification of ALL erases
both databases, TYPE erases just the type base, and ATOM erases just
the atom base (default: ALL).
- VDWOff
- type
turn off all REPEl interactions with atoms of this type.
type must already be defined by a NONB statement.
- VERBose
- produces a verbose listing of all atom-based parameters.
- nbonds-statement:==
-
- CDIERDIE
- specifies exclusive flags: constant
dielectric (Coulomb's law) or
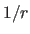 -dependent
dielectric (Eq. 4.16). RDIE may
only be used in combination with VSWItch, SWITch, and REPEl=0.
CDIE may be used in combination with VSWItch, SHIFt, and REPEl=0
or in combination with TRUNcation and REPEl=0 (default: CDIE).
-dependent
dielectric (Eq. 4.16). RDIE may
only be used in combination with VSWItch, SWITch, and REPEl=0.
CDIE may be used in combination with VSWItch, SHIFt, and REPEl=0
or in combination with TRUNcation and REPEl=0 (default: CDIE).
- CTOFNB=real
- specifies the distance
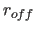 at which the
switching function or shifting function
forces the nonbonded energy to zero (Eqs. 4.8,
4.16) (default: 7.5 Å).
at which the
switching function or shifting function
forces the nonbonded energy to zero (Eqs. 4.8,
4.16) (default: 7.5 Å).
- CTONNB=real
- specifies the distance
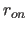 at
which the switching
function becomes effective (Eq. 4.8) (default: 6.5 Å).
at
which the switching
function becomes effective (Eq. 4.8) (default: 6.5 Å).
- CUTNb=real
- specifies the nonbonded
interaction cutoff
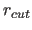 for
the nonbonded list generation (default: 8.5 Å).
for
the nonbonded list generation (default: 8.5 Å).
- E14Fac=real
- specifies the factor
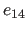 for the special
1-4 electrostatic interactions (Eq. 4.17)
(default: 1.0).
for the special
1-4 electrostatic interactions (Eq. 4.17)
(default: 1.0).
- EPS=real
- specifies the dielectric
constant (Eq. 4.16) (default: 1.0).
- GROUp ATOM
- specifies exclusive flags: group by group or atom by atom cutoff for nonbonded list generation (default: ATOM).
- INHIbit=real
- specifies the distance
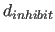 (Eq. 4.7) between two atoms
below which the van der Waals potential
(Eq. 4.8) is truncated (default: 0.25 Å).
(Eq. 4.7) between two atoms
below which the van der Waals potential
(Eq. 4.8) is truncated (default: 0.25 Å).
- IREXponent=integer
- specifies the exponent
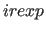 for the repel function (Eq. 4.8)
(default: 2).
for the repel function (Eq. 4.8)
(default: 2).
- NBXMod=
- Exclusion list options:
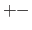 1
1- no nonbonded exclusions, that is, all nonbonded interactions are computed regardless of covalent bonds.
- 2
- excludes nonbonded interactions between bonded atoms.
- 3
- excludes nonbonded interactions between bonded atoms and atoms that are bonded to a common third atom.
- 4
- excludes nonbonded interactions between bonded atoms, atoms that are bonded to a common third atom, and or atoms that are connected to each other through three bonds.
- 5
- same as (+-3), but the 1-4 nonbonded interactions
are computed using the 1-4 Lennard-Jones
parameters and the electrostatic scale factor
(Eqs. 4.17 and 4.18).
- RCONst=real
- specifies the energy constant
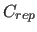 for
the repel function (Eq. 4.8)
(default: 100.0).
for
the repel function (Eq. 4.8)
(default: 100.0).
- REPEl=real
- specifies
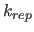 : if 0, this option
turns on the repel function (Eq. 4.8) and turns off the
electrostatic energy. specifies the factor by which
to multiply the van der Waals radius
: if 0, this option
turns on the repel function (Eq. 4.8) and turns off the
electrostatic energy. specifies the factor by which
to multiply the van der Waals radius 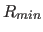 (default: 0).
(default: 0).
- REXPonent=integer
- specifies the exponent
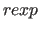 for the repel function (Eq. 4.8) (default: 2).
for the repel function (Eq. 4.8) (default: 2).
- SWItchSHIFt
- specifies exclusive flags: electrostatic switching or shifting. SWITch may only be used in combination with RDIE, VSWItch, and REPEl=0. SHIFt may only be used in combination with CDIE, VSWItch, and REPEl=0 (default: SHIFt).
- TOLErance=real
- specifies the distance that any atom
is allowed to move before the nonbonded list gets
updated. Note: if switching or shifting functions
are used, the program expects
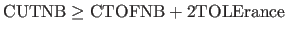 . In this
way the nonbonded energy is independent of the update frequency.
For the REPEl option, CUTNB and TOLErance should be chosen
such that
. In this
way the nonbonded energy is independent of the update frequency.
For the REPEl option, CUTNB and TOLErance should be chosen
such that
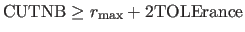 ,
where
,
where 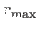 is the maximum van der Waals radius.
TOLErance has no influence on the TRUNcation option.
(default: 0.5 Å).
is the maximum van der Waals radius.
TOLErance has no influence on the TRUNcation option.
(default: 0.5 Å).
- TRUNcation
- turns off switching or shifting; i.e., the nonbonded energy functions are “truncated" at CUTNb regardless of the values of CTONNB and CTOFNB. All nonbonded energy terms that are included in the current nonbonded list are computed. May only be used in combination with CDIE. Note: in general, the nonbonded energy will not be conserved before and after nonbonded list updates when using TRUNcation. (default: inactive).
- VSWItch
- turns on van der Waals switching. May only be used in combination with RDIE, SWITch, and REPEl=0 or in combination with CDIE, SHIFt, and REPEl=0 (default: active).
- WMIN=real
- specifies the threshold distance for close contact warnings, i.e., a warning is issued when a pair of atoms gets closer than this distance unless the nonbonded interaction is excluded by the NBXMod option (default: 1.5 Å).
- CDIE
- hbonds-statement:==
-
- AAEX=real
- specifies the exponential for the
angle term between the hydrogen, acceptor and acceptor antecedent
atoms (Eq. 4.25) (default: 2).
- ACCEptor=logical
- is a flag indicating whether to include the acceptor antecedent angular term in Eq. 4.25 (default: TRUE).
- ACUT=real
- specifies the angular cutoff
for the angle between acceptor, hydrogen and donor atoms
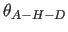 (Eq. 4.25).
(Eq. 4.25). 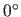 corresponds to a linear hydrogen bond
(default:
corresponds to a linear hydrogen bond
(default: 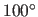 ).
).
- AEXP=real
- specifies the attractive exponential
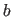 (Eq. 4.25) (default: 4).
(Eq. 4.25) (default: 4).
- AON=real
- specifies
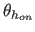 , a switching function
parameter for the hydrogen bond angle (Eq. 4.25)
(default:
, a switching function
parameter for the hydrogen bond angle (Eq. 4.25)
(default: 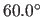 ).
).
- AOFF=real
- specifies
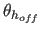 ,
a switching function
parameter for the hydrogen bond angle (Eq. 4.25)
(default:
,
a switching function
parameter for the hydrogen bond angle (Eq. 4.25)
(default: 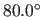 ).
).
- DCUT=real
- specifies
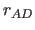 , the heavy atom
donor to heavy atom acceptor cutoff (Eq. 4.25)
(default: 7.5).
, the heavy atom
donor to heavy atom acceptor cutoff (Eq. 4.25)
(default: 7.5).
- DOFF=real
- specifies
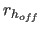 ,
a switching function parameter for the hydrogen bond
distance (Eq. 4.25) (default: 6.5).
,
a switching function parameter for the hydrogen bond
distance (Eq. 4.25) (default: 6.5).
- DON=real
- specifies
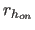 , a
switching function parameter for the hydrogen bond distance
(Eq. 4.25) (default: 5.5).
, a
switching function parameter for the hydrogen bond distance
(Eq. 4.25) (default: 5.5).
- HAEX=real
- specifies the exponential for the
angle term between the donor, hydrogen, and acceptor atoms,
(Eq. 4.25) (default: 4).
- PRINt=logical
- control printing of hydrogen bond neighbor information whenever the neighbor list is updated (default: TRUE).
- REXP=real
- specifies the repulsive exponential
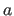 (Eq. 4.25) (default: 6).
(Eq. 4.25) (default: 6).
- TOLErance=real
- specifies how far atoms are allowed
to move before the hydrogen-bond list gets updated.
Note: the program expects
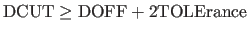 . In this
way the hydrogen-bonded
energy is independent of the update frequency (default: 0.5).
. In this
way the hydrogen-bonded
energy is independent of the update frequency (default: 0.5).
- AAEX=
Xplor-NIH 2026-07-10