
| bit | Xplor-NIH | VMD-XPLOR |
|---|
|
| Xplor-NIH home Documentation |
Next: Electron Density Map File Up: Computation of Electron Density Previous: Computation of Electron Density
Syntax
- MAP
- {
 fiber_refin-map-statement
fiber_refin-map-statement } END This
is a fiber_refin-statement. Computation takes place as
soon as the END statement is issued.
} END This
is a fiber_refin-statement. Computation takes place as
soon as the END statement is issued.
- fiber_refin-map-statement:==
-
- OUTPut=filename
- Name of the map file (default: standard output).
- GRID=real
- Sets grid size of the electron density map.
- AUTOmatic=logical
- indicates whether automatic scaling is performed. The
electron density map will have an average of zero and a
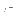 of one (default: true).
of one (default: true).
- EXTEnd=MOLEcule
 SUBUnit BOX
SUBUnit BOX - defines boundary type for the electron density map (default: MOLEcule). In MOLEcule type, the map will cover the selected atoms of the molecule. In SUBUnit type, the whole subunit is written. In BOX type, the user can explicitly specify the boundaries of a cubic box in orthogonal Å.
- CUSHion=real
- Specifies additional “cushioning” around the molecule in Å. Only for EXTEnd=MOLEcule mode (default: 2.0 Å).
- SELEction=selection
- Selects atoms for EXTEnd=MOLEcule mode. The map will be written such that it covers the selected atoms (default: all).
- XMIN
- sets lower x boundary of the BOX type electron density map in orthogonal Å (default: 0.0)
- XMAX
- sets upper x boundary of the BOX type electron density map in orthogonal Å (default: 1.0)
- YMIN
- sets lower y boundary of the BOX type electron density map in orthogonal Å (default: 0.0)
- YMAX
- sets upper y boundary of the BOX type electron density map in orthogonal Å (default: 1.0)
- ZMIN
- sets lower z boundary of the BOX type electron density map in orthogonal Å (default: 0.0)
- ZMAX
- sets upper z boundary of the BOX type electron density map in orthogonal Å (default: 1.0)
- OUTPut=