
| bit | Xplor-NIH | VMD-XPLOR |
|---|
|
| Xplor-NIH home Documentation |
Next: Template Structure Up: XPLOR Interface Manual Previous: CPU and Memory Requirements
NMR Structure Determination
X-PLOR can be used to determine
and refine solution NMR structures based on interproton
distance estimates, coupling constants measurements, and other
information, such as known hydrogen-bonding patterns.
Figure 38.1 presents an overview of distance-based NMR structure determination. There are several alternative methods possible in X-PLOR: full-structure distance geometry, substructure distance geometry, and ab initio simulated annealing starting from template structures or random coordinates. The choice of protocol depends on the desired efficiency and sampling of conformational space. Generally, for all well-determined NMR structures, the pathway indicated by black lines should be followed, i.e., substructure embedding and regularization, full-structure SA regularization, and SA refinement. To sample alternative conformations that cannot be sampled with the simple distance geometry substructure approach, the full structure distance geometry protocol with metrization or the ab initio simulated annealing protocol starting from a template structure should be used. [For a discussion of sampling characteristics of distance geometry, see Kuszewski, Nilges, and Brünger (1992).] Both options are clearly more time-consuming than substructure distance geometry. For very large structures (larger than 2000 atoms), full-structure distance geometry with metrization is likely to push the memory and CPU limits of most computers. The ab initio simulated annealing protocol (“sa.inp") provides an important alternative here. The simulated annealing protocol starting from random coordinates (“random.inp") has been included to provide yet another possibility. It shows that simulated annealing is so powerful that it can convert a random array of atoms into a well defined structure through NOE distance restraints. Structures obtained by this protocol have to be regularized by the “dgsa.inp" protocol.
(For back-calculations and complete matrix relaxation refinement, see Chapter 39.)
Structure determination of extended polypeptides or DNA/RNA double strands is usually underdetermined. In particular, the overall shape or bend of the molecule is a free parameter. Thus, neither distance geometry nor ab initio simulated annealing will produce unique structures. The problem can be avoided by including additional restraints, such as repulsive distance restraints between appropriately chosen phosphate groups in the case of RNA or DNA, or by starting the refinement process from ideal conformations. In the latter case, one should use the “refine.inp" or “refine_gentle.inp" protocols to refine the starting coordinates. It is not necessary to run “generate_template.inp" and the subsequent distance geometry or simulated annealing stages. For example, A- and B-type conformations of DNA or RNA double strands could be obtained from other programs (presently, X-PLOR does not provide a facility to produce ideal DNA conformations). These starting coordinates should be refined and then the convergence of the refined coordinates should be assessed.
If disjoint subunits are present in the molecular structure, distances or covalent bonds need to be defined between them. Otherwise, the relative separation between the subunits is unknown, causing the distance geometry algorithm to place the subunits far apart from each other. This may trigger a message about unknown atomic coordinates if certain atoms have coordinates larger than 9999.9 Å.
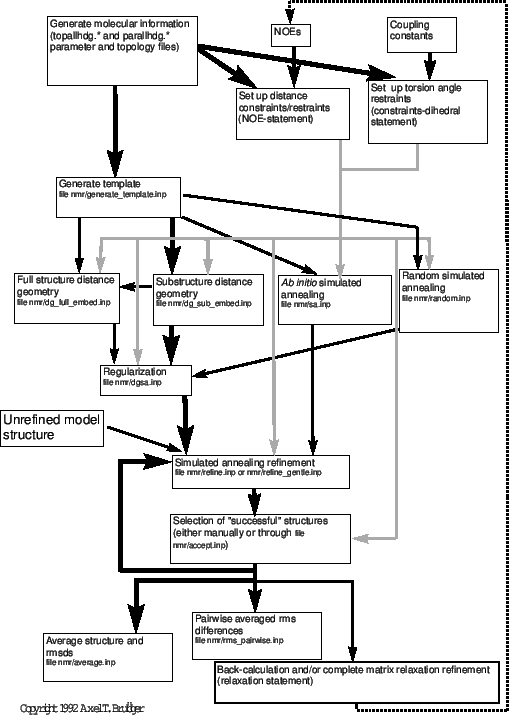
The first step of NMR structure determination using X-PLOR involves providing the program with the information it needs about the molecular structure, NOE-derived distance bounds, and coupling-constant-derived dihedral angle restraints. The molecular information of the macromolecule has to be generated using the all-hydrogen force fields “topallhdg.pro", “parallhdg.pro" for proteins and “topallhdg.dna", “parallhdg.dna" for nucleic acids. The generation of the molecular structure should proceed as described in Section 3.7. Note that one has to include all known disulfide bridges in the case of protein structures in the molecular structure generation. Special handling of disulfide bridges and other covalent links occurs in a number of protocols described in this section, and all examples assume that the disulfide bridges have been properly defined. Contrary to earlier versions of X-PLOR, no internal coordinate information is required for any of the NMR protocols.
The “topallhdg.*" force fields for NMR structure determination (Section 3.6) contain improper angles to define the chirality of chiral and prochiral centers. If a coordinate file is used that is not generated by X-PLOR, the local chirality of the coordinates must match the definition used in X-PLOR's parameter sets. The user should check this by computing the energy of the structure with the energy statement (see Section 4.6). The chirality of the chiral and prochiral centers is dependent on the hydrogen naming scheme. An example of how to change the naming convention is provided in the file “topallhdg.convert" in the “toppar" directory (cf. Section 3.6.10). It is clearly imperative that the naming convention used for stereospecific assignments must match the one used by the particular force field.
The setup of NOE-derived distance bounds is described in Chapter 20, and the setup of coupling-constants-derived dihedral angle restraints is described in Section 7.2. For ambiguous or unresolved NOE assignments, distance bounds should be entered that are appropriate for pseudoatoms; i.e., in all examples described in this section, the center-averaging method is used for the NOE restraint function. For example, in the case of a methyl group, the distance is restrained to the geometric center of the methyl hydrogens. This also applies to the distance geometry applications, because the X-PLOR distance geometry routine automatically computes a pseudoatom correction to obtain appropriate distance bounds for each of the methyl group protons individually.
The final value of the REPEl value (cf. Section 3.2.1) as employed in the simulated annealing and refinement protocols, 0.75, may seem a little small (see also Section 3.6.10). It has been arrived at by inspection of Ramachandran plots of unrestrained polypeptides (Nilges, unpublished results). The allowed region of conformational space is similar to the “generous" region described by Morris et al. (1992). With this value, the final van der Waals energy should be very small. Larger values of REPEl up to 0.8 are also acceptable. The general effect of increasing REPEL will be a reduction of the rms distribution of an ensemble of coordinates, and an increase in the final potential energy.
Subsections
- Template Structure
- Options: Distance Geometry, Ab Initio SA, or Random SA
- Distance Geometry
- Test for the Correct Enantiomer
- Options: Full-Structure Embedding or SA
- SA-Regularization of DG-Structures
- Full-Structure Distance Geometry
- Ab Initio SA Starting from the Template
- Random Simulated Annealing
- Simulated Annealing Refinement
- Acceptance of Refined NMR Structures
- Average Structure and Rmsds
- Pairwise Rmsds
- Time-Average Refinement
- CPU Time Requirements
Next: Template Structure Up: XPLOR Interface Manual Previous: CPU and Memory Requirements Contents Index