
| bit | Xplor-NIH | VMD-XPLOR |
|---|
|
| Xplor-NIH home Documentation |
Next: Non-crystallographic Symmetry Interactions Up: Nonbonded Energy Terms Previous: Intramolecular Interactions
Crystallographic Symmetry Interactions
Crystallographic symmetry interactions (e.g., packing interactions)
between the
molecule(s) located in the asymmetric unit and all symmetry-related
molecules
surrounding the asymmetric unit are computed by
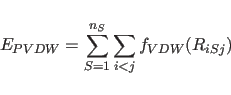 |
(4.19) |
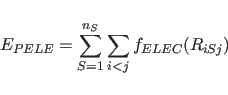 |
(4.20) |
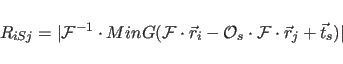
The function
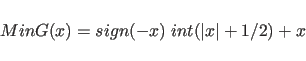
Xplor-NIH 2026-07-10