
| bit | Xplor-NIH | VMD-XPLOR |
|---|
|
| Xplor-NIH home Documentation |
Next: Requirements Up: Crystallographic Diffraction Data Previous: Orthogonalization Convention
Syntax of the Xrefin Statement
The xrefin statement is used to read structure factors,
symmetry operators, atomic-form factors, unit cell parameters, etc.
It also branches into several other statements that allow
one to manipulate structure factors, refine certain parameters,
carry out translation or rotation searches, compute
solvent masks, or compute
electron density maps.
Manipulations
of structure factors are carried out for the selected
reflections. The selection of reflections is accomplished
by the RESOlution and FWINdow statements. Atoms contributing
to the structure factor ![]() are selected through
the SELEction and SCATter statements.
are selected through
the SELEction and SCATter statements.
- XREFin {
 xrefin-statement
xrefin-statement } END
} END - is invoked from the main level of XPLOR.
- xrefin-statement:==
-
- A=real
- specifies
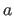 of unit cell (default: 1.0 Å).
of unit cell (default: 1.0 Å).
- ALPHa=real
- specifies
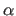 of unit cell
(default: 90
of unit cell
(default: 90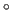 ).
).
- B=real
- specifies
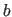 of unit cell (default: 1.0 Å).
of unit cell (default: 1.0 Å).
- BETA=real
- specifies
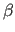 of unit cell
(default: 90).
of unit cell
(default: 90).
- C=real
- specifies
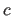 of unit cell (default: 1.0 Å).
of unit cell (default: 1.0 Å).
- DO
- xrefin-do-statement manipulates structure
factors (see Section 13.5).
- EXPAnd
- expands selected reflections to P1; i.e., the crystallographic symmetry operators and the Hermitian symmetry operator (if HERMitian=TRUE) are applied to the selected reflections. Multiple entries that emerge during this process are discarded. Multiple application of EXPAnd is possible. The second time around, it does not have any effect unless new data are added or more symmetry operators are added.
- FFK=real
- sets
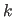 in Eq. 13.1 to the specified value.
If FFK is set to 0, is automatically
determined by Eq. 13.6.
Automatic scaling also declares symbols ($FFK, $TEST_FFK)
that contain the values of for the working set and the
test set, respectively
(default: 0, i.e., automatic scaling).
in Eq. 13.1 to the specified value.
If FFK is set to 0, is automatically
determined by Eq. 13.6.
Automatic scaling also declares symbols ($FFK, $TEST_FFK)
that contain the values of for the working set and the
test set, respectively
(default: 0, i.e., automatic scaling).
- FFT
- { FFT-statement } END specifies
parameters for the FFT method.
- FWINdow
- real real sets
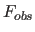 amplitude
limits for the selection of reflections. One of the real
values is the upper value; the other one is the lower value;
it does not matter whether the upper or the lower
limit comes first (default: FWINdow 0 100000).
amplitude
limits for the selection of reflections. One of the real
values is the upper value; the other one is the lower value;
it does not matter whether the upper or the lower
limit comes first (default: FWINdow 0 100000).
- GAMMa=real
- specifies
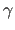 of unit cell (default: 90).
of unit cell (default: 90).
- GENErate
- complements the current reflections to yield a full asymmetric unit of reflections for the specified resolution range. If no current reflections are present, a full asymmetric unit is generated. The new Fobs are set to 1 except for systematic absences, in which case Fobs is set to 0. Fcalc, Fpart are set to 0; weight, sigma, and FOM are set to 1. Action is taken as soon as this statement is issued.
- HERMitian=logical
- specifies whether Friedel mates are identical (HERMitian=TRUE) or different from each other (HERMitian=FALSE). Thus, for anomalous scattering data, one has to set HERMitian=FALSE and specify the imaginary scattering components for the appropriate atoms (default: TRUE).
- LOOKup=logical
- is a flag indicating whether to use lookup tables for the direct summation method and the FFT method (default: TRUE). Turn off the lookup tables if problems with minimization procedures are encountered.
- MAP
- { xrefin-map-statement } END
computes electron density maps (see
Section 16.1).
- MBINs=integer
- specifies the number of bins
for the
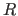 value analysis (PRINt R), phase difference
analysis (PRINt PHASe), data completeness
analysis (PRINt COMPleteness), and
computation of normalized structure factors
(Es) (default: 8).
value analysis (PRINt R), phase difference
analysis (PRINt PHASe), data completeness
analysis (PRINt COMPleteness), and
computation of normalized structure factors
(Es) (default: 8).
- METHod=
- DIREct
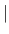 FFT specifies choice of
method to compute
FFT specifies choice of
method to compute 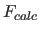 (default: FFT).
(default: FFT).
- NREFlections=integer
- is a required parameter that allocates space for the reflections. It has to be greater than or equal to the actual number of reflections (default: 200).
- OPTImize
- BFACtor
{ xrefin-optimize-bfactor-statement }
END optimizes individual (restrained) isotropic B-factors (see
Section 14.4).
- OPTImize
- GROUp { xrefin-optimize-group-statement }
END optimizes group B-factors and/or occupancies (see
Section 14.3).
- OPTImize
- OVERall
{ xrefin-optimize-overall-statement }
END optimizes an overall isotropic or anisotropic B-factor (see Section 14.2). - PRINt COMPleteness
- prints the ratio of the number of observed reflections to the number of theoretically observable reflections (“completeness," printed as a percentage). The analysis is carried out as a function of resolution. The overall completeness is stored in the symbol $COMPLETENESS. If the data are partitioned into a test set and a working set, a completeness analysis is also carried out for the test reflections (see Chapter 17). In this case, the completeness for the test reflections (TEST=1) is stored in the symbol $TEST COMPLETENESS, and the completeness for the working set is stored in $COMPLETENESS. This statement also produces a listing that can be plotted by a Mathematica script.
- PRINt PHASe
- prints the average phase difference
as a function of resolution for the selected
reflections and stores the overall phase
difference in the symbol $DPHI. Phases differences
are weighted with the WEIGht array (see Section 13.5).
If a figure-of-merit
weighted average is required, the
user should issue a “DO (WEIGHT=MAX(0,FOM))" statement;
this will overwrite the existing weights.
If the data are partitioned into a test set and a working
set, a phase difference analysis for the test
reflections is also carried out (see Chapter
17). The “free" phase difference
is stored
in the symbol $TEST DPHI. Note that
no prior update of is performed, and thus the
user must issue an UPDAte statement if the
array is undefined or not well defined.
This statement
also produces a listing that can be plotted by a
Mathematica script.
- PRINt R
- prints the value
as a function of resolution for the selected
reflections and stores the
overall value in
the symbol $R.
If the data are partitioned into a test set and a working
set, a free
value analysis is also carried out (see Chapter
17). The free value is stored
in the symbol $TEST R. Note that
no prior update of is performed. Thus,
the user must update with the UPDAte
statement if the array is undefined or
if an energy calculation (e.g., energy minimization or
molecular dynamics) was carried out previously.
In the latter case, the array is
filled with the values of the derivatives of the
target function. This statement
also produces a listing that can be plotted by a
Mathematica script.
- PRINt TARGet
- prints the value
of
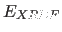 (Eq. 13.1). If
the residual or the AB vector-residual
is chosen, the value is stored in the symbol $R.
If targets are chosen that contain a correlation coefficient,
its value is stored in the symbol $CORR.
If the data are partitioned into a test and a working
set (see Chapter 17), the corresponding values
for the test set are stored in the symbols $TEST R
and $TEST CORR.
Note that
no prior update of is performed, and thus the
user must issue an UPDAte statement if the
array is undefined or not well defined.
(Eq. 13.1). If
the residual or the AB vector-residual
is chosen, the value is stored in the symbol $R.
If targets are chosen that contain a correlation coefficient,
its value is stored in the symbol $CORR.
If the data are partitioned into a test and a working
set (see Chapter 17), the corresponding values
for the test set are stored in the symbols $TEST R
and $TEST CORR.
Note that
no prior update of is performed, and thus the
user must issue an UPDAte statement if the
array is undefined or not well defined.
- PRINt WILSon
- makes a Wilson plot
(Wilson, 1949; Main, 1975; Rogers, 1965).
The overall scale factor and B-factor
are obtained by a least-squares fit of
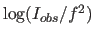 vs.
vs. 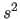 . The result is stored in symbols
$BFACTOR, $SCALE, and $TEST_BFACTOR, $TEST_SCALE for
the working and test sets, respectively. This statement
also produces a listing that can be plotted by a
Mathematica script.
. The result is stored in symbols
$BFACTOR, $SCALE, and $TEST_BFACTOR, $TEST_SCALE for
the working and test sets, respectively. This statement
also produces a listing that can be plotted by a
Mathematica script.
- REDUce
- decreases selected reflections to an asymmetric unit. In the case of multiple entries, the first entry is kept and the duplicates are discarded; i.e., no data averaging is performed.
- REFLection
- { xrefin-reflection-statement } END
initiates reading or
merging of diffraction data (see Section 13.4).
- RESEt
- erases the current xrefin database, i.e., the atomic-form factors, symmetry operators, reflections, and unit cell parameters.
- RESOLution
- real real sets resolution limits
in Å for the selection of reflections. One of the
real values is the high resolution limit, and the other one
is the low resolution limit; it does not matter whether the high
or the low resolution limit comes first.
One of the limits can be set to the string “INFInity",
e.g., “RESOlution INFInity 3".
This statement
will then include the 0,0,0 reflection in all
calculations (default: RESOlution
10. 3. ).
- SCATter
- selection real real
real real real real real
real real [ IMAGinary real ]
adds an atomic-form factor specification to the xrefin
database. The statement specifies the coefficients
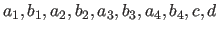 (Eq. 13.10)
for the selected atoms
(default: none). An atom
will contribute to the structure factors only if it has
been selected in one SCATter statement and if it
has been selected in the SELEction statement.
Care should be taken not to produce an overlapping
definition of atom selections; e.g., if there is a
ca ion, one
should exclude it from the atomic-form factor definition for
carbon atoms. The optional IMAGinary parameter should be used
for anomalous scatterers.
“SCATter RESEt" will erase the existing atomic
form factor entries.
(Eq. 13.10)
for the selected atoms
(default: none). An atom
will contribute to the structure factors only if it has
been selected in one SCATter statement and if it
has been selected in the SELEction statement.
Care should be taken not to produce an overlapping
definition of atom selections; e.g., if there is a
ca ion, one
should exclude it from the atomic-form factor definition for
carbon atoms. The optional IMAGinary parameter should be used
for anomalous scatterers.
“SCATter RESEt" will erase the existing atomic
form factor entries.
- SEARch
- ROTAtion { xrefin-search-rotation-statement
} END is a translation search (see
Section 19.3).
- SEARch
- TRANslation
{ xrefin-search-translation-statement } END
is a rotation search (see
Section 19.3).
- SELEction=
- selection selects atoms that will
be used in the next structure factor calculation (default: (ALL) ).
An atom
will contribute to the structure factor only if it has
been selected in one SCATter statement and
has been selected in the SELEction statement.
The selection remains active until a new SELEction statement
is issued.
- SOLMask
- { xrefin-solmask-statement } END
computes a solvent mask (see Section 13.7).
- SYMMetry=symmetry-operator
- adds a new symmetry operator
(
 ) to the xrefin database. The
notation is the same as in the International
Tables for Crystallography (Hahn, 1987),
e.g.,
) to the xrefin database. The
notation is the same as in the International
Tables for Crystallography (Hahn, 1987),
e.g.,
Multiple entries specify the space group. A listing of the symmetry operators of all crystallographic space groups is stored in file “symlib.sym" in the “symmetry" directory. In the case of centered space groups, all symmetry operators have to be specified; otherwise symmetry images will be missing for the packing interactions. Normally, this redundant specification of symmetry operators represents only a small increase in CPU time. The option “SYMMetry RESEt" will erase the existing symmetry operators (default: symmetry=(x,y,z)).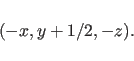
(13.11) - TARGet=
- RESIdual AB F1F1 F2F2 E1E1
E2E2 PACKing specifies choice of the target
function; see Eq. 13.1 (default: RESIdual).
- TOLErance=real
- specifies the maximum amount
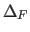 (in Å) by which
any atomic coordinate can deviate from the position when
was last computed during molecular
dynamics, energy minimization, or energy calculations.
If TOLErance is exceeded, and its
derivatives with respect to atomic parameters
are recomputed (default: 0.5 Å).
(in Å) by which
any atomic coordinate can deviate from the position when
was last computed during molecular
dynamics, energy minimization, or energy calculations.
If TOLErance is exceeded, and its
derivatives with respect to atomic parameters
are recomputed (default: 0.5 Å).
- UPDAte
- computes for selected reflections
using the current atomic model and atomic-form factors.
- WA=real
- specifies the overall weight factor
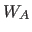 in
Eq. 13.1 (default: 1).
in
Eq. 13.1 (default: 1).
- WP=real
- specifies the overall weight factor
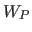 for the phase term
for the phase term 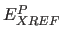 in Eq. 13.1
(default: 0).
in Eq. 13.1
(default: 0).
- WRITe
- REFLection { write-reflection-statement }
END
writes the specified xrefin properties, such as FOBS or
FCALC, of all selected reflections to a specified output file
(see Section 13.4).
- A=
- FFT-statement:==
-
- AVOID=integer
- facilitates avoidance of the specified integer as
a factor in the
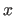 and
and 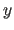 physical dimension
of the 3-d electron density matrix in order
to avoid memory conflicts on certain supercomputers
(default: machine dependent, set automatically).
physical dimension
of the 3-d electron density matrix in order
to avoid memory conflicts on certain supercomputers
(default: machine dependent, set automatically).
- BASE=integer
- specifies the minimum prime allowed in FFT dimensions (default: machine dependent, set automatically).
- BSCALefactor=real
- sets the artificial temperature factor
to minimize aliasing effects (default: 20.0 Å
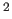 ).
).
- ELIMit=real
- defines an atomic “radius" for the electron density calculation by specifying the ratio of the atomic-form factor at zero and at the radius of the atom in natural logarithmic units (default: 7.00). The electron density outside the radius is set to zero.
- GRIDsize=real
- specifies the grid size relative to the high resolution limit (default: 0.33, which corresponds to 1/3 of the high resolution limit).
- MEMOry=integer
- indirectly determines the factorization of the FFT by specifying the maximum memory allocation allowed for the electron density matrix in units of complex words (default: 500000).
- PRIMe=integer
- specifies the maximum prime allowed for FFT dimensions (default: machine dependent, set automatically).
- AVOID=
Subsections
Next: Requirements Up: Crystallographic Diffraction Data Previous: Orthogonalization Convention Contents Index