
| bit | Xplor-NIH | VMD-XPLOR |
|---|
|
| Xplor-NIH home Documentation |
Next: Requirements Up: Rigid-Body Coordinate Space Previous: Iteration
Syntax of the
Dynamics Rigid Statement
This statement sets up various parameters for rigid-body dynamics and
then executes the specified number of dynamics steps.
Parts of the molecule that are not specified in any
GROUp statement will remain fixed. The constraints fix
statement (Section 8.1)
has no influence on rigid-body molecular dynamics (except
for the default group selection).
- DYNAmics RIGId
- {
 dynamics-rigid-statement
dynamics-rigid-statement } END
is invoked from the main level of X-PLOR.
} END
is invoked from the main level of X-PLOR.
- dynamics-rigid-statement :==
-
- ASCIi=logical
- writes a formatted (ASCII)
coordinate trajectory file if ASCIi is TRUE;
if ASCIi
is FALSE, an unformatted (binary) file is written.
In the former case,
a scale factor
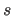 and an offset
and an offset 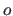 are applied to the
coordinates
are applied to the
coordinates 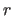 (i.e.,
(i.e.,
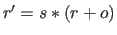 ); the result is converted to an
integer number and written to the
ASCII file using the specified format.
If a hexadecimal format is specified,
care should be taken to ensure that the
application of the scale factor and the offset
results in positive
coordinates
during
the whole course of the molecular dynamics simulation.
Otherwise, FORTRAN format runtime errors will occur.
); the result is converted to an
integer number and written to the
ASCII file using the specified format.
If a hexadecimal format is specified,
care should be taken to ensure that the
application of the scale factor and the offset
results in positive
coordinates
during
the whole course of the molecular dynamics simulation.
Otherwise, FORTRAN format runtime errors will occur.
- CSEL=selection
- selects atoms that are written to the coordinate trajectory file (default: all atoms except those fixed by the constraints fix statement).
- DT=real
- assigns the time step
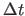 value for the finite-difference integration in psec
(default: 0.001 psec).
value for the finite-difference integration in psec
(default: 0.001 psec).
- DYNMODE=string
- selects from three possible rigid-body dynamics modes: FREE, LANGevin, or TCOUpling (default: FREE).
- FORMat=string
- provides a FORTRAN format for the ASCIi= TRUE option (default: 12Z.6, hexadecimal).
- GROUp=selection
- selects atoms that will form a rigid group. Multiple specification of GROUps will define multiple rigid groups. The group selections have to be disjoint (default: one group consisting of all atoms except those that have been fixed by the constraints fix statement; the default is overwritten as soon as a GROUp statement is issued).
- NPRInt=integer
- determines the frequency with which the energy is printed in standard output (default: 1, i.e., at every dynamics step).
- NSAVC=integer
- determines the frequency with which the coordinates are written to the coordinate trajectory file (default: 0).
- NSAVV=integer
- determines the frequency with which the velocities are written to the velocity trajectory file (default: 0).
- NSTEp=integer
- determines the number of steps (default: 100).
- NTRFrq=integer
- determines the update frequency to remove the overall center-of-mass translation and rotation of all “free" atoms (default: 0).
- OFFSet=real
- provides an offset for the ASCIi=TRUE
option (default: 800).
- SCALe=real
- provides a scale factor for the
ASCIi=TRUE option (default: 10000).
- TBATh=real
- specifies the temperature of the heatbath or the target temperature of the temperature-coupling method.
- TCOUpling=logical
- denotes the Berendsen temperature-coupling method, which works in conjunction with TBATh (default: FALSE).
- TRAJectory=file
- names coordinate trajectory file (default: none).
- VASCii=logical
- writes a formatted (ASCII) velocity
trajectory file if VASCii is TRUE; if VASCii
is FALSE, an unformatted (binary) velocity
trajectory file is written.
In the former case,
a scale factor and an offset are applied to the
velocities (i.e.,
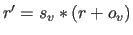 ); the result is converted to an
integer number and written to the
ASCII file using the specified format.
If a hexadecimal format is specified,
care should be taken to ensure that the
application of the scale factor
); the result is converted to an
integer number and written to the
ASCII file using the specified format.
If a hexadecimal format is specified,
care should be taken to ensure that the
application of the scale factor 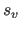 and the offset
and the offset 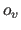 results in positive
velocities
during
the whole course of the molecular dynamics simulation.
Otherwise, FORTRAN format runtime errors will occur.
results in positive
velocities
during
the whole course of the molecular dynamics simulation.
Otherwise, FORTRAN format runtime errors will occur.
- VELOcity=file
- names the velocity trajectory file (default: none).
- VFORmat=string
- is the FORTRAN format of velocity trajectory file for the VASCii= TRUE option (default: 12Z.6, hexadecimal).
- VOFFset=real
- provides an offset for the VASCii= TRUE
option (default: 300).
- VSCAle=real
- provides a scale factor for the
VASCii=TRUE option (default: 10000).
- VSEL=selection
- selects atoms that are written to the velocity trajectory file (default: all atoms except those fixed by the constraints fix statement).
- ASCIi=
Xplor-NIH 2026-07-10