
| bit | Xplor-NIH | VMD-XPLOR |
|---|
|
| Xplor-NIH home Documentation |
Next: Initialization Up: Molecular Dynamics Previous: Example: Run Langevin Dynamics
Rigid-Body Coordinate Space
Rigid-body dynamics solves Newton's equations of
motion for rigid
collections of atoms (Goldstein, 1980).
Atoms
are collected into rigid groups, the motion of which is determined by
summing the forces acting on all of a group's elements and integrating
the rigid-body equations of motion.
The atomic masses
After completion of the molecular dynamics calculation, the
partial energy terms for the last molecular dynamics
step are stored in the appropriate
symbols.
The name of the symbols is given by
$![]() energy-term
energy-term![]() (see Section
4.5). The overall energy (Eq. 4.1)
is stored in the symbol
$ENER; the rms gradient is stored in $GRAD. The value of
the second energy function (Eq. 4.26) is
returned in the symbol $PERT.
In addition, the following symbols are declared:
$TEMP, $TOTE, and $TOTK,
which are respectively the temperature,
total energy, and kinetic energy.
(see Section
4.5). The overall energy (Eq. 4.1)
is stored in the symbol
$ENER; the rms gradient is stored in $GRAD. The value of
the second energy function (Eq. 4.26) is
returned in the symbol $PERT.
In addition, the following symbols are declared:
$TEMP, $TOTE, and $TOTK,
which are respectively the temperature,
total energy, and kinetic energy.
X-PLOR's implementation of rigid-body molecular dynamics
follows the algorithm described by Head-Gordon and Brooks (1991).
The algorithm treats each group as a continuous mass distribution
located at the center-of-mass position defined by
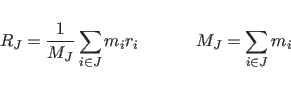 |
(11.11) |
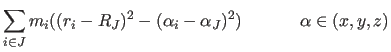 |
(11.12) | ||
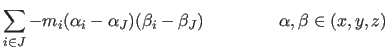 |
(11.13) |
Here the index J labels the rigid bodies, and the summation index
Subsections Xplor-NIH 2026-07-10