
| bit | Xplor-NIH | VMD-XPLOR |
|---|
|
| Xplor-NIH home Documentation |
Next: Syntax Up: Topology, Parameters and Molecular Previous: Example: Topology of a
Parameter Statement
The parameter
statement specifies various constants for the conformational
and nonbonded energy terms (Section 4.1) and for
a number of other X-PLOR applications, such as distance
geometry (Section 37).
Figure 3.1 illustrates the data flow for the parameters. X-PLOR stores and manipulates the parameter information in two forms: “type-based" and “atom-based". A type-based parameter is characterized by the chemical types of the atoms involved; an atom-based parameter is characterized by the individual atoms involved. The chemical types of the atoms are specified in the topology statement (Section 3.1.1) and can be manipulated with the vector statement (Section 2.16). The chemical types usually map chemically equivalent atoms to the same type; e.g., all carbonyl oxygen atoms are mapped to the type “O". This implies that there are usually many more atoms in the molecular structure than there are atom types. The use of atom types can thus reduce the number of parameters that need to be specified for a molecular structure. However, this convenience could pose a problem when the chemical environment of a particular atom induces distortions of the expected “ideal" geometry for the corresponding atom type. It would not necessarily be a good idea to introduce a new atom type, as the number of atom types is limited. To get around this problem, X-PLOR supports atom-based parameter information. This implies that, at least in principle, one could have a different parameter for each interaction between particular atoms. Atom-based parameters are possible for bonds, bond angles, dihedral angles, improper angles, and van der Waals interactions. They are presently not supported for the explicit hydrogen-bonding energy term.
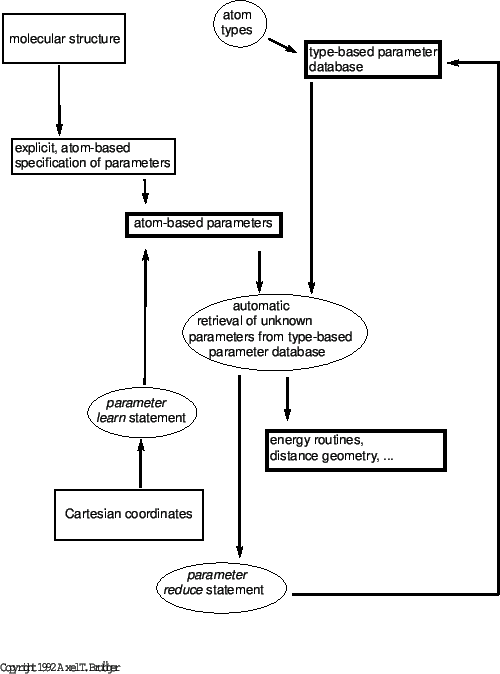
X-PLOR allows one to mix atom-based and type-based parameters. Atom-based parameters always take precedence over type-based parameters. The atom-based parameter information can be obtained through appropriate explicit parameter statements, such as
bond ( name c* ) ( name n* ) 500. TOKENor through the Cartesian coordinates in the main coordinate set in combination with the learn statement. This atom-based information is augmented with the type-based information each time an X-PLOR application requests it; e.g., the call to an energy routine triggers an operation that retrieves all parameters not set by atom-based information from the type-based parameter database. In this way either atom-based parameters, type-based parameters, or a combination of both can be used. Atom-based parameters can be changed or added at any time during program execution. Type-based parameters cannot be manipulated, but additional entries can be added or the database erased and reinitialized. The mapping of atoms to chemical types can be manipulated by applying the vector statement to the chemical atom property (Section 2.16). All these changes will take effect the next time the particular parameter is requested by an X-PLOR application.
The learn statement can be used to generate atom-based
parameters for interactions involving selected atoms. Parameters
can be learned from a single coordinate set or from an
ensemble of coordinates. Equilibrium constants
for bonds, bond angles,
dihedral angles, or improper angles can be learned. Energy
constants can be obtained from the standard deviation
of the equilibrium parameters around their mean. Clearly,
this is only possible when an ensemble average of
coordinate sets is available. Improper and dihedral angles
are learned under the assumption that the
periodicity ![]() is zero (Eq. 4.6) regardless of
any typed-based default periodicity.
It should be noted that the generation
of a template structure (Section 38.1)
is essentially the inverse
of the learn statement: it creates a molecular conformation
with ideal geometry from the parameters.
is zero (Eq. 4.6) regardless of
any typed-based default periodicity.
It should be noted that the generation
of a template structure (Section 38.1)
is essentially the inverse
of the learn statement: it creates a molecular conformation
with ideal geometry from the parameters.
The reduce statement tries to map the atom-based parameters into type-based parameter information using the chemical atom property. The redundancy of this process implies that the parameter information needs to be averaged. Existing type-based parameters can be overwritten or stay untouched, depending on the specified option. The newly created type-based parameter set can be written to a parameter file for use with other X-PLOR protocols. Only bond, bond angle, dihedral angle, and improper angle parameters can be learned or reduced. Energy constants can be obtained either by averaging the atom-based values or by calculating the standard deviation of the equilibrium values from their mean for a particular instance of type-based parameters.
The parameter specifications are insensitive to the order in which the atoms are specified; e.g.,
bond a b 10.0 1.0and
bond b a 10.0 1.0are seen as identical parameter entries by X-PLOR.
The constants of the parameter statement
are dimensioned such that Å is the
unit of length, kcal mole![]() is the unit of energy, the atomic mass unit
is the unit of mass, and the charge of one electron is the unit of
electric charge.
is the unit of energy, the atomic mass unit
is the unit of mass, and the charge of one electron is the unit of
electric charge.
Obtaining empirical parameters for macromolecules is a difficult task. To describe the development of parameters is beyond the scope of this book. X-PLOR includes a number of parameter files for proteins, nucleic acids, and other biological macromolecules (Section 3.6). Fortunately, structure determination and refinement of crystallographic or NMR-derived structures are usually not very sensitive to the exact choice of energy constants. The learn statement provides a convenient tool to obtain the equilibrium geometry for chemical compounds that are not included in the parameter files. The equilibrium parameters can be obtained from known Cartesian coordinates of the compounds. The energy constants can be set to uniform values or can be derived from a statistical analysis of the structures of several related compounds. Note that the learn statement does not actually create new interaction terms (bonds, bond angles, improper angles, dihedral angles) for the molecular structure. It simply assigns parameters for interaction terms that are already defined in the molecular structure. The creation of new interaction terms is described in Section 3.8.
Subsections Xplor-NIH 2026-07-10