
| bit | Xplor-NIH | VMD-XPLOR |
|---|
|
| Xplor-NIH home Documentation |
Next: Orthogonalization Convention Up: Crystallographic Diffraction Data Previous: Crystallographic Diffraction Data
Crystallographic Target Functions
X-PLOR provides several possibilities for the
effective energy ![\begin{displaymath}
E_{XREF} = \left\{ \begin{array}{l}
W_A/N_A \sum_{\vec{h}} ...
...}(\vec{h})^2]) \\
W_A (1- {\rm Pack} )
\end{array} \right.
\end{displaymath}](img385.png)
for the first choice in Eq. 13.1, the unweighted vector
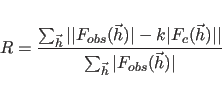
for the second choice, or the various correlation coefficients for the third to sixth choices. The
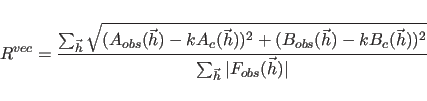
The selection of reflections
is accomplished by the RESOlution and FWINdow statements (see below).
“Corr" is defined through
![\begin{displaymath}
{\rm Corr}[x \;, \; y] = \frac{<xy-<x><y»}{\sqrt{<x^2-<x>^2> \; <y^2-<y>^2>}}
\end{displaymath}](img399.png) |
(13.4) |
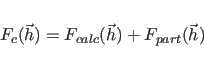 |
(13.5) |
The normalized structure factors
(![]() s) are computed from
the structure factors (
s) are computed from
the structure factors (![]() s) by averaging the
s) by averaging the ![]() s in equal
reciprocal volume shells within the specified resolution
limits. The number of shells is specified by
MBINs.
s in equal
reciprocal volume shells within the specified resolution
limits. The number of shells is specified by
MBINs.
The purpose of the normalization factor
![]() (first and second choice in Eq. 13.1)
is to make the weight
(first and second choice in Eq. 13.1)
is to make the weight ![]() approximately independent of the resolution
range during SA-refinement.
approximately independent of the resolution
range during SA-refinement. ![]() has been set to
has been set to
![]() .
The scale
factor
.
The scale
factor ![]() in Eq. 13.1 is set to
in Eq. 13.1 is set to
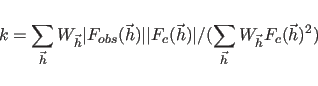
The term ![]() represents phase restraints
if
represents phase restraints
if ![]() is set
to a nonzero number.
is set
to a nonzero number.
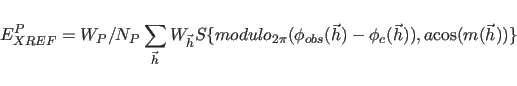
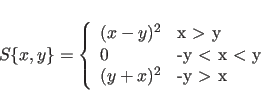 |
(13.8) |
The structure factors (
![]() ) of the atomic model
are given by
) of the atomic model
are given by
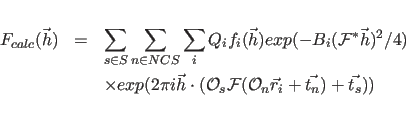
The first sum extends over all symmetry operators
The constants
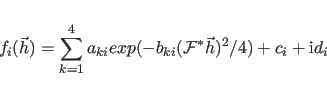
An approximation is used to reduce the computational requirements when
multiple evaluations of Eq. 13.1 are required.
The approximation involves not computing
![]() and its
first derivatives at every dynamics or minimization step. The first
derivatives are kept constant until any atom has moved by more than
and its
first derivatives at every dynamics or minimization step. The first
derivatives are kept constant until any atom has moved by more than
![]() (TOLErance in xrefin statement)
relative to the position at which the derivatives were last
computed. At that point, all derivatives are updated. Typically,
(TOLErance in xrefin statement)
relative to the position at which the derivatives were last
computed. At that point, all derivatives are updated. Typically,
![]() is set to 0.2 Å for dynamics
and to 0-0.05 Å for minimization.
is set to 0.2 Å for dynamics
and to 0-0.05 Å for minimization.
The PACKing target
is defined for evaluating the likelihood of
packing arrangements of the search model and its symmetry mates in the
crystal (Hendrickson and Ward, 1976).
A finite grid that
covers the unit cell of the crystal is generated. The grid
size is specified through the GRID parameter in the
xrefin FFT statement.
All grid points
are marked that are within
the van der Waals radii around any atom of the search model and its
symmetry mates. The number of marked grid points
represents the union of the molecular spaces of the search model
and its symmetry mates. Maximization of the union of molecular
spaces is equivalent to minimization of the overlap. Thus,
an optimally packed structure has a maximum of the packing
function. “Pack" in Eq. 13.1
contains the ratio of the number of marked
grid points to the total number of grid points in the unit cell.
For instance, a value of 0.6 means 40% solvent contents.
![]() is then set to 0.4 if
is then set to 0.4 if ![]() .
.
For further reading on the crystallographic target functions in X-PLOR, see Brünger (1988,1990,1989).
Xplor-NIH 2026-07-10