
| bit | Xplor-NIH | VMD-XPLOR |
|---|
|
| Xplor-NIH home Documentation |
Next: NCS Restraints Up: XPLOR Interface Manual Previous: Cross-validation: The Free Value
Non-crystallographic Symmetry
Non-crystallographic symmetry (NCS) has been introduced into X-PLOR in two
different ways. In the first, NCS-related atoms are restrained in their
average positions by means of an effective energy term analogous to the
covalent
bond energy.
This is the method used, for example, by the refinement program PROLSQ
(Hendrickson, 1985).
The user specifies groups of NCS-equivalent
atoms. A least-squares superposition of these atoms onto a reference set
(taken as the first set defined) is computed, and the average x,y,z for
each atom is taken. Each atom is then restrained according to the energy
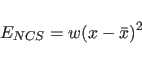 |
(18.1) |
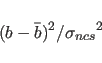 |
(18.2) |
The second way assumes that all NCS-related
molecules are strictly identical
.
Specification of the coordinate
trajsformations defining the NCS is given by the
NCS strict statement. All energy calculations, such as minimization
and dynamics, are performed on the unique (protomer) atoms only. For the
X-ray terms, the NCS coordinate transformations are applied to the input
protomer to generate F![]() s and derivatives for the entire
crystallographic asymmetric unit, and the derivatives are transformed back
to the protomer coordinates to apply an NCS-averaged force.
s and derivatives for the entire
crystallographic asymmetric unit, and the derivatives are transformed back
to the protomer coordinates to apply an NCS-averaged force.
For the case of strict NCS, the nonbonded interactions among NCS-related protomers can be treated in a manner analogous to crystal symmetry contacts: the operators defining the NCS relations are used to compute the nonbonded interactions between the protomer being refined and its neighbors. By the assumptions of strict symmetry, the crystal lattice contacts are ignored in this option. For structures in which some of the internal symmetry is non-crystallographic and some is crystallographic (e.g., icosahedral viruses), additional operators besides those needed for structure factor calculations (i.e., a subset of the crystallographic operators) can be input to complete the specification of the nonbonded interactions arising from the internal symmetry. The interprotomer nonbonded interactions are turned on by setting the packing flags PVDW and PELE (Section 4.5; these energies are used for the internal symmetry interactions only, as the crystal contacts are automatically shut off).
The NCS strict statement and the NCS restraints statement are independent; i.e., the two options can be used together. In particular, NCS restraints within a protomer can be refined under the STRIct option.
There is a third way to impose symmetry restraints on
a dimer based on distance differences (Section
20.8). This alternative allows
one to impose exact twofold symmetry on a dimer without
specifying the actual operation between the monomers; i.e., the
separation between the monomers will be a self-adjusting parameter.
Subsections
Next: NCS Restraints Up: XPLOR Interface Manual Previous: Cross-validation: The Free Value Contents Index