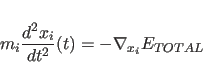An official website of the United States government
Here’s how you know
Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
)or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
Molecular dynamics consists of solving Newton's equations of
motion
(11.1)
where the index runs through all free (i.e.,
not fixed) atoms and the
gradient
is derived from the
X-PLOR energy function (Chapter 4).
Presently, X-PLOR provides the option to
solve Eq. 11.1 in Cartesian coordinate space
(Section 11.1) or rigid-body coordinate space
(Section 11.2). Molecular dynamics
simulations can be stored as trajectory files.
Input format, output format, manipulations, and
analysis of trajectory
files are described in Section 11.